Silin
Zhong Lab | Plant Functional Genomics CUHK
钟思林 实验室 | 植物功能基因组学 香港中文大学
School of Life Sciences
The Chinese University of Hong Kong
Tel: 3943 6280
Home Research Jobs ENCODE data browser
https://www.nature.com/articles/s41467-022-35438-4

https://pubmed.ncbi.nlm.nih.gov/35605196/
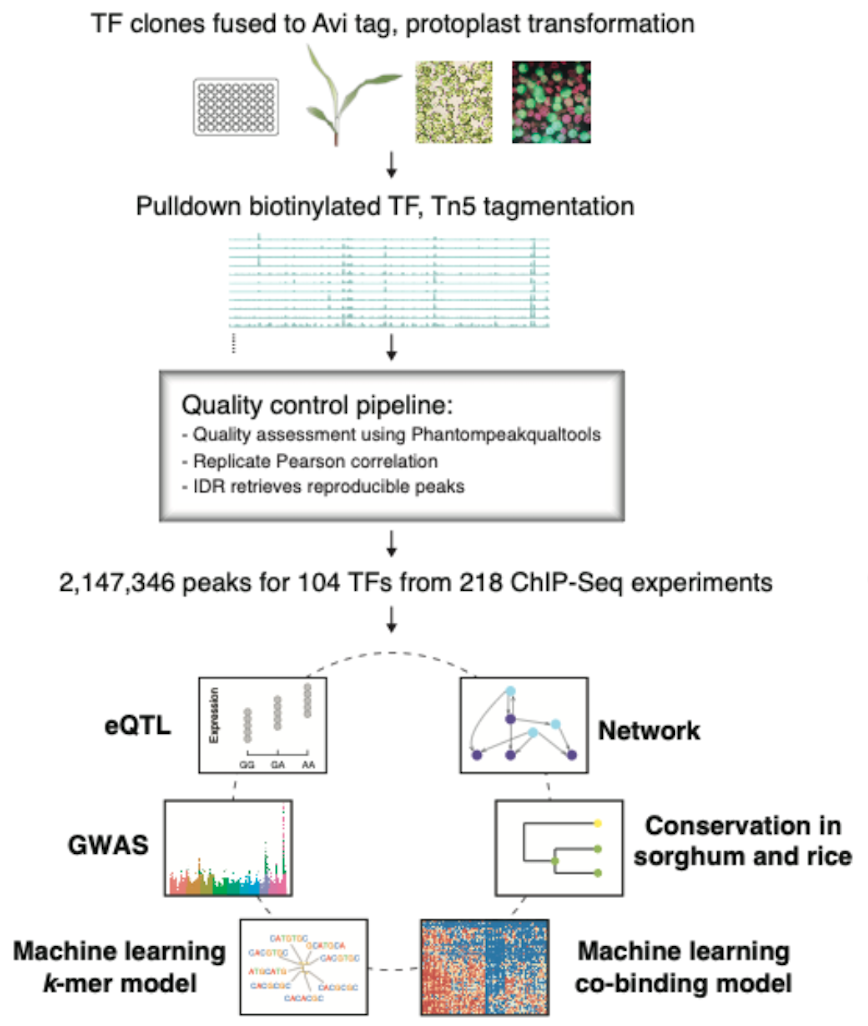
https://www.nature.com/articles/s41467-020-18832-8
2018 | The FruitENCODE project
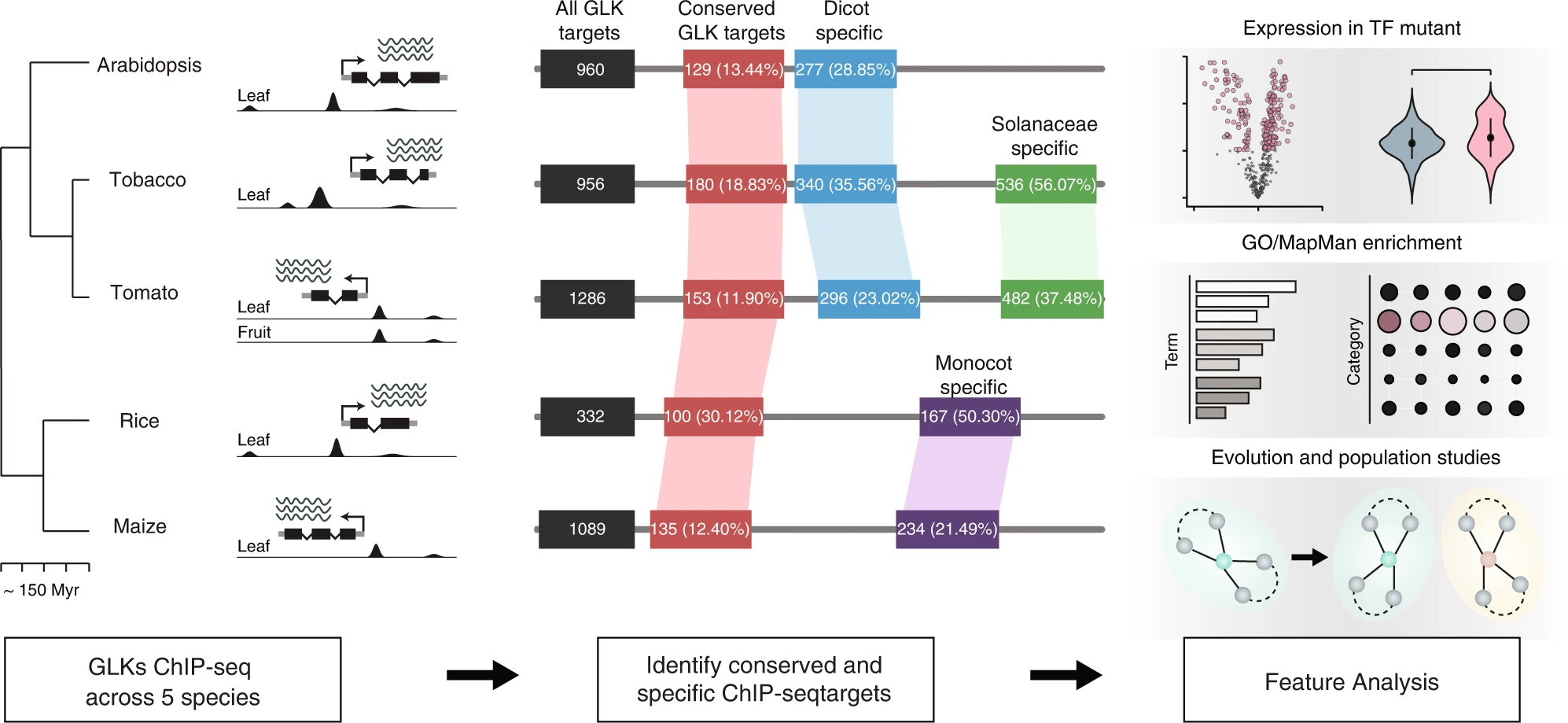
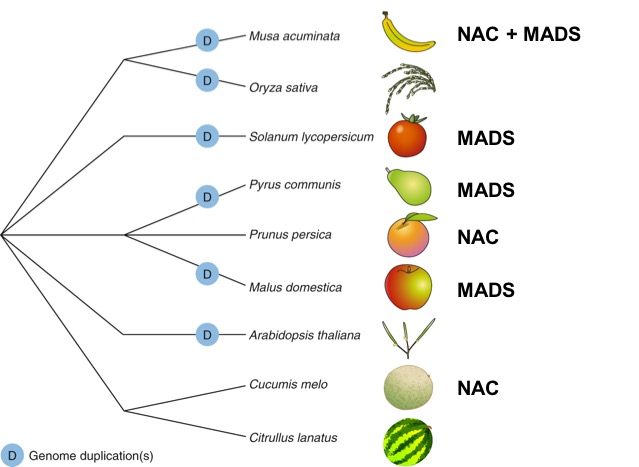
Analysis of the fruitENCODE data reveals three types of transcriptional feedback circuits controlling ethylene-dependent fruit ripening. These circuits are evolved from senescence or floral organ identity pathways in the ancestral angiosperms either by neofunctionalisation or repurposing pre-existing genes. The epigenome, H3K27me3 in particular, has played a conserved role in restricting ripening genes and their orthologues in dry and ethylene-independent fleshy fruits.

2017 | Large crop genome chromatin 3D organization revealed by Hi-C analysis
2017 MP cross-species comparison
2019 JIPB Tissue-specific Hi-C
2020 JXB Review

https://www.nature.com/articles/nbt.2462